

Science: Catalytic active sites, where do they come from?
2024-08-02
Understanding the nature of active sites on catalytic surfaces is central to understanding and improving catalysis. Low-index metal surfaces are commonly used models of supported metal catalysts, especially in computational studies, but have limitations in describing active sites under real reaction conditions. STM studies have shown that when molecules are adsorbed on a low refractive index surface, sub-nanometer metal clusters with more low-coordination atoms exposed can be formed in situ on the low refractive index surface. Compared with nanoparticles (NPs) with a diameter of> 2 nm, this metal cluster exhibits different catalytic and electronic properties and dynamic structural flux, and NPs can also be recombined during the reaction. The adsorption-induced cluster formation implies the dynamic nature of the catalytic surface and suggests that the active sites formed under the actual relevant reaction conditions may be essentially different from those in the extended surface or the original NP model usually used for thermal and electrocatalytic mechanism studies. Although highly dispersed small metal clusters are usually prepared by controlled synthesis techniques, the formation of clusters induced by adsorbents on bulk crystals under realistic reaction conditions has not been explicitly included in previous studies.

existing problems
Understanding the nature of active sites on catalytic surfaces is central to understanding and improving catalysis.Low-index metal surfaces are commonly used models of supported metal catalysts, especially in computational studies, but have limitations in describing active sites under real reaction conditions.STM studies have shown that when molecules are adsorbed on a low refractive index surface, sub-nanometer metal clusters with more low-coordination atoms exposed can be formed in situ on the low refractive index surface. Compared with nanoparticles (NPs) with a diameter of> 2 nm, this metal cluster exhibits different catalytic and electronic properties and dynamic structural flux, and NPs can also be recombined during the reaction.The adsorption-induced cluster formation implies the dynamic nature of the catalytic surface, and shows that the active sites formed under the actual relevant reaction conditions may be related to the original on the extended surface or usually used for thermal catalysis and electrocatalytic mechanism research.The active site in the NP model is fundamentally different.Although highly dispersed small metal clusters are usually prepared by controlled synthesis techniques, under realistic reaction conditions,The induction of cluster formation on bulk crystals by adsorbents has not been explicitly included in previous studies.
Solution ideas
Based on this,University of Wisconsin-MadisonManos Mavrikakis等UseEnergetics from DFT calculations,Development of an effective framework for predicting adsorption-induced cluster formation. It is first discussed how to relate the adsorption-induced clustering effect to an easily calculated energy parameter. Then, it is demonstrated how this approach leads to predictions consistent with experimental results, as well as its limitations. Further proposed a comprehensive database including8 transition metals and 18 adsorbents. Subsequently, a case study of CO-induced cluster formation on Cu(111) was performed using Kinetic Monte Carlo (KMC) simulations.The paper is based on 《Formation of active sites on transition metals through reaction-driven migration of surface atoms》题发表在ScienceOn.
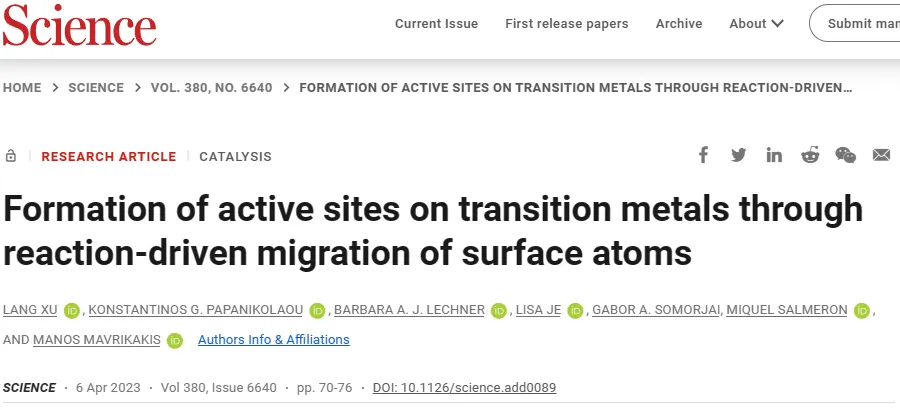
Bare surface and adsorptionTheoretical modeling of CO surfaces
The formation of adsorbate-induced clusters is mainly a three-step process(Figure1A):(i)The ejection of step edge atoms or junction atoms leads to the formation of adatoms on adjacent terraces,(ii) Adatom diffusion, and (iii) Adatom aggregation to form sub-nano clusters. Since the first step of the process requires breaking the surface metal-Metal bond, so the energy cost of the atomic bomb must be overcome to carry out these three-step process.Study defines the adatom formation energy(Eform) as a preliminary screening system for descriptors that can demonstrate clustering. The procedure for evaluating the formation energy of attached atoms involves the use of two infinitely separated plate models(Figure1B-C), one of which is the source where the surface atoms are ejected, and the other is the site where the atoms fall after being ejected from the first plate.
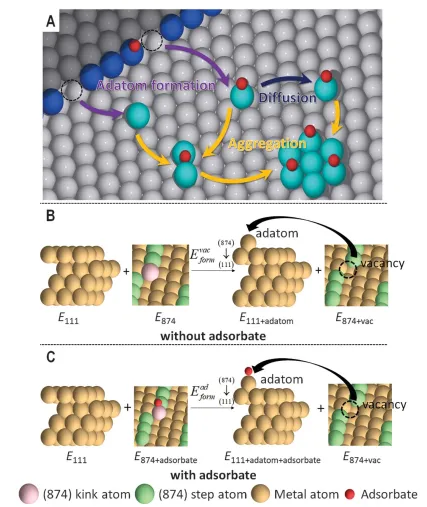
Figure1: Definition of the formation energy of adhesion atoms
Study considers three possible ejection sources: (I) kink location; (ii) regular, defect-free step edges; and (iii)(111) terrace.DueEformis a thermodynamic parameter that does not fully capture the kinetics of adatom formation, which is determined by the energy barrier. Therefore, the calculatedEformvalue can be considered as a lower bound on the activation energy barrier of adatom formation. First, the vacuum(Figure2A) and the presence of CO (Figure2B), CO is a common reactive species in catalytic applications. In the absence of adsorbents, in the presence of eight face-centered cubic (fcc) transition metals (Ag, Cu, Au, Pd, Ni, Rh, Pt, and Ir)Not Refactored(111) Adsorbed atoms formed by (874) kinked atom ejection on the surface are always the most likely of the above three ejection sources.Au and Pt are two exceptions, indicatingBulk metal hardness cannot fully capture the energy of the atomic bomb.InOn Ag(874), Cu(874) and Au(874), CO promotes the formation of adatoms, while on the other five metals, CO inhibits the formation of adatoms.InE on 8 metal surfacesformValue(Figure 2B) spanning from 0.42 to 2.11 eV, demonstrating the huge difference in cluster formation potentials in the periodic table. Based onFigure2BPrediction of Results and Normal Temperature and High Pressure-STM (HP-STM) results are consistent, indicating thatCO can induce cluster formation on Cu(111), but not on Rh(111).. The stress energy near the step and surface dislocations, as well as the interaction between the steps, will also greatly affect the energetics of the step atom bomb. Explicit modeling of high coverage of adjacent surfaces lends itself to a more detailed study of a particular system. Still,This model can be used as an adhesive atom and/or indicators of cluster formation trends, suitable for large-scale screening purposes.
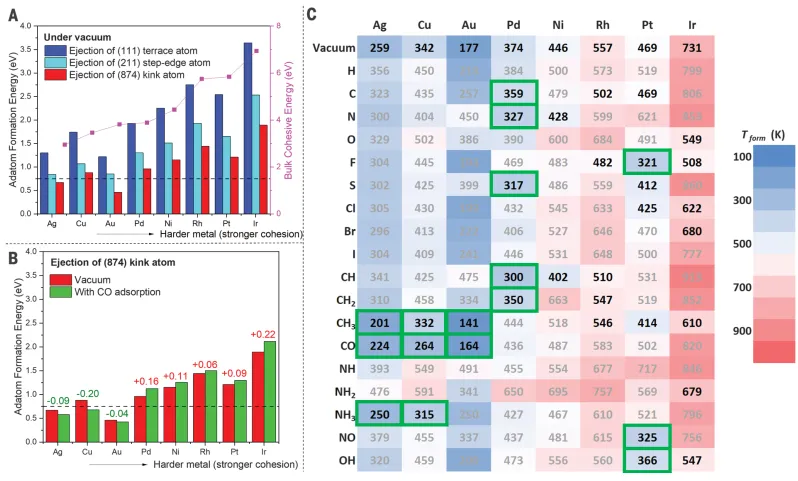
Figure 2: Atomic formation energy and temperature
Simulation of the effect of reaction intermediates on cluster formation
Extend the scope of study to a wide range of adsorption reaction intermediates(H, C, N, O, F, S, Cl, Br, I, CH, CH2, CH3It is equipment2, T/C3, NO and OH), these intermediates usually participate in heterogeneous catalysis.The formation of metal adatoms is always endothermic, regardless of the elemental nature of the adsorbent or metal, and the source face of the metal adatoms. The effect of the adsorbent on the formation of adatoms depends largely on its binding characteristics on the corresponding metal surface,No generalized adsorbent effect was observed with respect to the adsorbent promoting or inhibiting the formation of adatoms.
In order to establish a connection with realistic catalytic reaction conditions, the temperature at which these adsorption-induced adatom formation events may occur was estimated.(Figure2 c)。The possible occurrence of adsorbate-induced adatom formation at near ambient temperatures in the system was determined on the basis of two criteria: (I) The adsorbent should promote the formation of adsorbed atoms and (ii) the estimated adsorption formation temperature (Tformad) value below373 K(100℃). InIn the presence of CO, clusters are formed on Cu(111), indicating the potential correlation of CO-induced clusters with CO oxidation on Cu catalysts. To verify this hypothesis, Cu formed on Cu(111) was calculatednCO oxidation to CO on (n = 1~4) clusters2The activation energy barrier(Figure3A-B). in these Cu(111) loaded clusters,The Cu trimer (Cu3 +) exhibits the lowest transition state energy for CO oxidation, which is also smaller than the transition state energy value on any extended Cu crystal plane studied here.At room temperature, accordingDFT results, the activity of Cu3 + for CO oxidation is estimated to be ~ 2 × 10 higher than that of Cu(874)6This result proves thatPotential role of CO-induced Cu cluster sites in catalyzing CO oxidation.
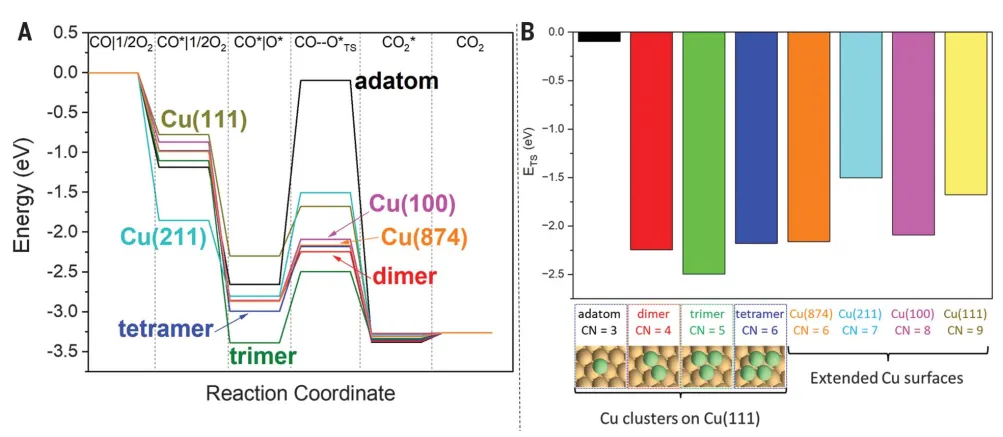
Figure 3: Cluster effect of Cu-catalyzed CO oxidation
KMC simulation of the formation of active sites on Cu surface
In order to explore the dynamics of atomic bomb and cluster formation process, the copper atoms from the preferred ejection source copper(874) The CO-induced Cu(111) cluster formation process caused by ejection in the knot.in differentThe activation energy barrier of kinked atomic bomb in Cu(874) under CO cover scales linearly with approximately one slope with ejection energy, indicating that Cu(874) is in a late transition state(Figure4A)。The ejection activation energy barrier is at most larger than the respective ejection energy0.02 eV. Therefore, use onlyThe kinetics of CO-induced Cu(874) atomic ejection can be well described by the thermochemical reaction of Cu(874). In addition, it is also determined that the CO coverage of Cu(874) kink and step sites has the greatest influence on the ejection dynamics. When the kink and step sites are completely covered by CO, the ejection dynamics are most likely to occur.studied in the following three cases, fromThe overall thermochemistry associated with the formation of small clusters of finite size on Cu(874) from the beginning of the Cu(111) kink atomic bomb: (I) vacuum,(ii) medium CO coverage,(iii) high CO coverageCover(Figure 4B). The presence of CO enhances the stability of the finally formed cluster, making the overall cluster formation process thermodynamically favorable. For the intermediate CO cover, corresponding to the saturated Cu step edge and kink site, the calculated activation energy barriers for adatom diffusion and adatom attachment on existing Cu5 + clusters on Cu(111) to form Cu6 + are 0.04 and 0.05 eV, respectively, while the atomic ejection barrier is 0.37 eV (Figure4C). At room temperature, the atomic bomb is at least 5 orders of magnitude slower than the diffusion and attachment processes, which confirms thatThe formation of the initial adatoms is rate-limiting for the entire cluster formation process, including the atomic bomb and cluster formation.In order to better relate our theoretical resultsExperimental observations of STM are linked by the energetics of cluster expansion to predict nanocluster formation on Cu by KMC simulations.When the shot energy decreasesAt 0.15 eV, we observed that at 0.3 torr CO exposure and room temperature, it is most consistent with the experimental STM measurement results (Figure 4D). observed formation of small clusters in the early stages of the simulation(<5 atoms). These clusters later grow to 20-50 atoms and decorate the CO molecules mainly in the peripheral positions of the clusters (Figure4D), similar to what was observed in the experiment.
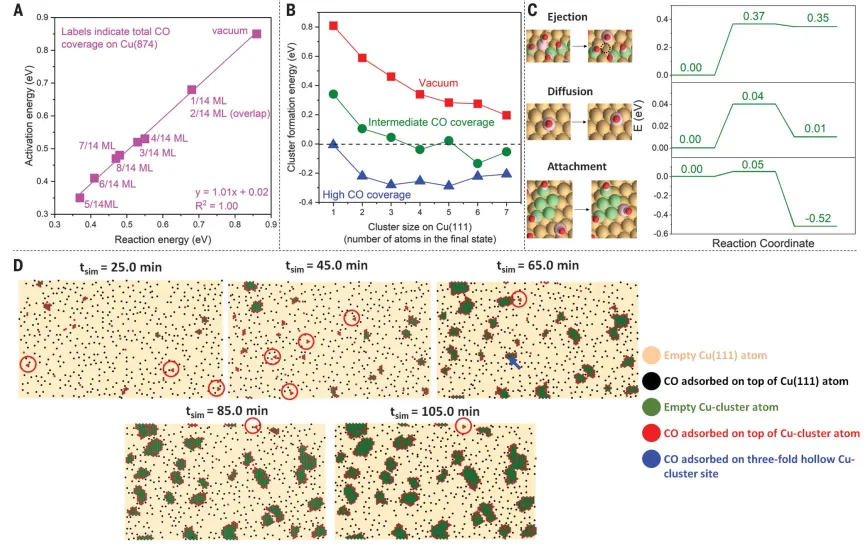
Figure 4: Due to the ejection of Cu(874) kink atoms, CO induces the formation of clusters on Cu(111)
Structural sensitivity of STM
Figure2A-B show that as the coordination number of the ejected atom decreases, the emission energy also decreases.In order to explore the role of defect sites and strain, the useThe effect of CO on Ni(111) single crystals exhibiting edge and screw dislocation defects is studied by STM.Two kinds of steps, straight-line and wavy-type steps, are observed with atomic heights of both~ 0.2 nm. Unexpectedly, it is observed that nickel adatoms and small clusters form even at a CO dose of 77 K(Figure5). At low CO doses, the wavy step edges are decorated with bright protrusions, while the straight step edges remain original (Figure5A). With further exposure of CO, the decoration of the undulating steps becomes denser and the number of clusters on the rungs increases (Figure5B). With the further increase of CO exposure, CO molecules are also decorated on each Ni-order atom (Figure5C). The change of step brightness shows that there is no participation of nickel atoms in the decoration of the straight step compared with the wavy step. In some of the smallest clusters (Figure5D), single protrusion, dimer and trimer species can be observed, which can be explained as one, two and three Ni atoms covered by CO molecules, respectively.In order to better understandNi attached atoms and cluster formation process, we in situ continuous exposure to ~ 4 x 10− 9The evolution of the surface was tracked when torr CO. Figure 5E-Fshowed that the cluster was first confined to the upper right terrace, but at longerThe CO exposure begins to cover the rest of the surface. As the CO coverage increases, some clusters are locked in place by the adsorbed CO molecules, and fewer streaks appear. The apparent background roughness increases, indicating that the adsorption of CO starts from the edge of the step and then diffuses to the terrace.

Figure 5:STM images showing the effect of CO exposure on defective Ni(111) surfaces
Summary
studies construct a framework for predicting adsorption-induced metal-Breaking of metallic bonds on solid surfaces and subsequent formation of clusters on these surfaces. Knowledge of the nature of the active site can serve as a starting point for screening catalytic systems with potential adsorption-induced clustering behavior.Importantly, the findings may reshape our understanding of catalytic reaction conditions for metalsUnderstanding the nature of active sites on NPs.The research framework shouldContribute to the design of more accurate active site models in theoretical studies, as well as to guide complex field experimental studies of more practically relevant catalytic systems.
References
https://www.science.org/doi/10.1126/science.add0089
Lang Xu et al. Formation of active sites on transition metals through reaction-driven migration of surface atoms.Science380, 70-76 (2023).
DOI:10.1126/science.add0089

RELATED INFORMATION






Contact us
Website: www.amoycat.cn
Address: No. 66 Xinyuan South Road, Haicang District, Xiamen City, Fujian Province, China

Official account QR code